Over the years, our scientists have been tasked with a variety of complex LC-MS/MS studies. In their research, they worked to produce cost-saving procedures, deliver reproducible results, and employ automation technologies.
Here is an overview of the Bioanalytical study research presented at scientific conferences over the past four years. Click the links to view the full poster; including analysis, details, and conclusions.

1.) Prove Microsampling accuracy with LC-MS/MS
Meng Fang shares our microsampling study. Download the poster from the app or stop by booth 1255. #aaps2016 pic.twitter.com/p5jwzmBCxI
— Alliance Pharma (@Alliance_Phar) November 15, 2016
Mouse Pharmacokinetic Study of Ceftriaxone Using Mitra™ Microsampling Devices and LC-MS/MS
In this study, an experiment was designed to compare a serial blood sampling method using Mitra™ microsamplers with parallel blood sampling methods using both Mitra™ microsamplers and venipuncture.
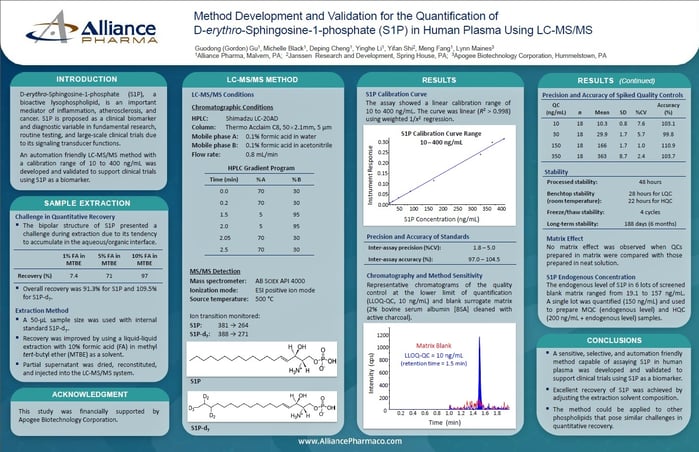
2.) Automate the work of quantifying S1P in Human Plasma using LC-MS/MS
Method Development and Validation for the Quantification of D-erythro-Sphingosine-1-phosphate (S1P) in Human Plasma Using LC-MS/MS
An automation friendly LC-MS/MS method with a calibration range of 10 to 400 ng/mL was developed and validated to support clinical trials using S1P as a biomarker.
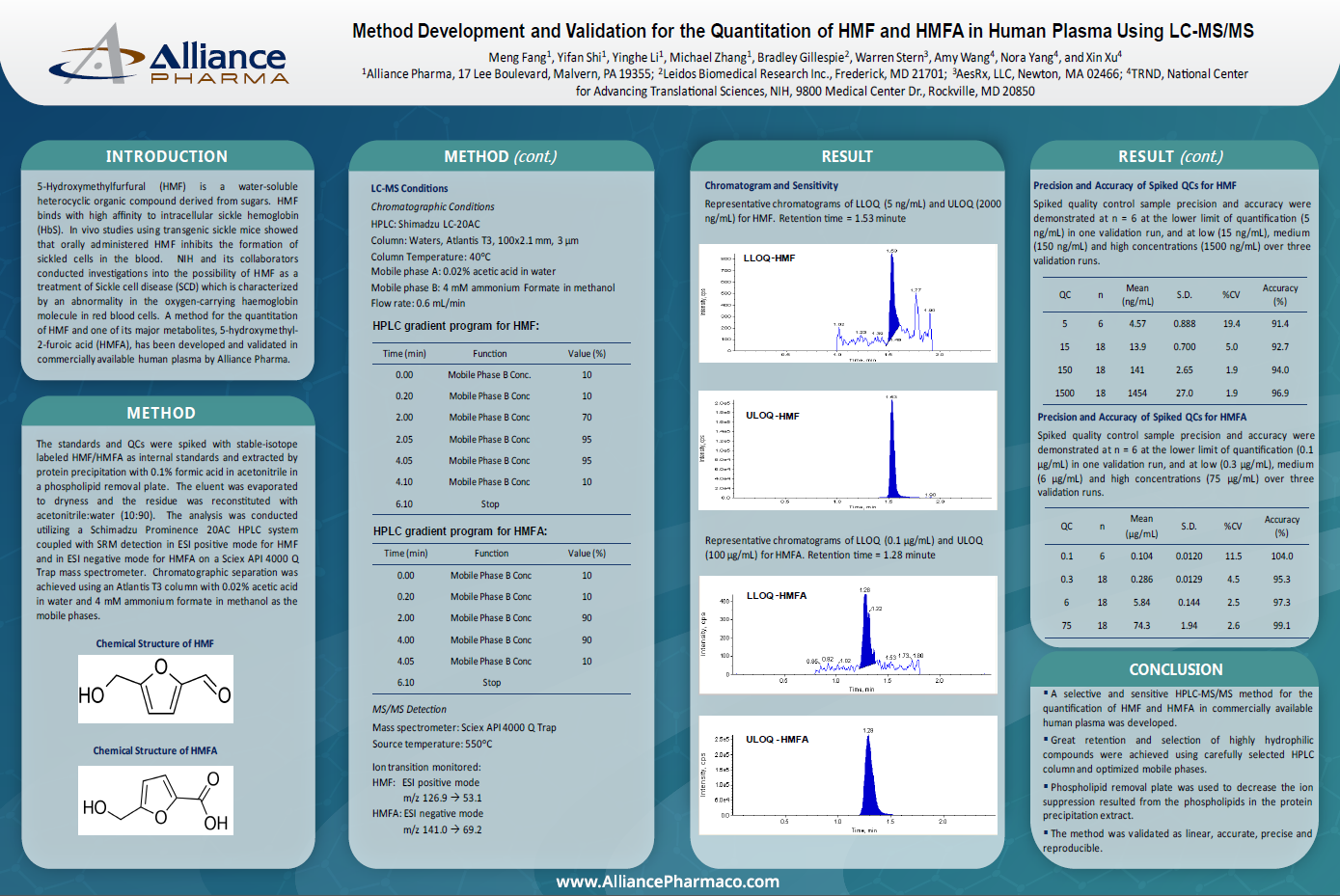
3.) Quantify HMF and HMFA in Human Plasma with LC-MS/MS
Method Development and Validation for the Quantitation of HMF and HMFA in Human Plasma Using LC-MS/MS
A method for the quantitation of HMF and one of its major metabolites, 5-hydroxymethyl-2-furoic acid (HMFA), has been developed and validated in commercially available human plasma by Alliance Pharma.

4.) Confirm stability with rapid and sensitive LC-MS/MS method
SHORTCUT to CYP Enzyme Activity Monitoring
A rapid and sensitive LC-MS/MS method was validated for cortisol and 6ß-HC analysis
in human urine. Method accuracy, precision, repeatability, selectivity, F/T stability, processed sample stability, bench-top stability, and long term stability have been confirmed.
5.) Evaluate drug-drug interactions during LC-MS/MS clinical studies
Quantification of 4b-Hydroxycholesterol and Cholesterol in Human Plasma Using LC-MS/MS
The plasma concentration ratio of 4b-hydroxycholesterol (4b-HC) to cholesterol has been recognized as a reliable marker for the assessment of Cytochrome P450 (CYP) 3A4 activity. It could be a valuable yet simple and cost effective side-product of a clinical study to evaluate CYP3A4-mediated drug-drug interactions.
6.) Support a Phase I clinical study with an LC-MS/MS method
Method Development and Validation for the Quantitation of ManNAc in Human Plasma Using HILIC LC-MS/MS
The purpose of this study was to develop and validate an LC-MS/MS method for assaying N-acetylmannosamine (ManNAc) in human plasma (K2EDTA) to support a phase I clinical study.
7.) Measure Goserelin in human plasma using LC-MS/MS
A Rapid and Sensitive Method for the Quantification of Goserelin in Human Plasma Using HPLC-MS/MS
The purpose of this study was to develop and validate a rapid and sensitive LC-MS/MS method for measuring Goserelin in human plasma (K2EDTA).

8.) Determine the quantification of 25-hydroxyvitamin D3 with an LC-MS/MS method
Quantification of 25-hydroxyvitamin D3 in Rat Serum Using Derivatization to Enhance LC-MS/MS Sensitivity
In this study, a sensitive and robust LC-MS/MS method was developed and validated for the determination of 25-OHVD3 in rat serum.
9.) Quantify Mercaptoethanol through LC-MS/MS analysis
Quantification of 2-Mercaptoethanol in Bulk Drug Substance by LC-MS/MS
Here, we report a LC-MS/MS method that utilizes derivatization with picolinic acid to quantitatively analyze residual mercaptoethanol and has been successfully used in biopharmaceutical manufacture.