D-erythro-Sphingosine-1-phosphate(S1P), a bioactive lysophospholipid, is an important mediator of inflammation, atherosclerosis, and cancer. S1P is proposed as a clinical biomarker and diagnostic variable in fundamental research, routine testing, and large-scale clinical trials due to its signaling transducer functions.
An automation friendly LC-MS/MS method with a calibration range of 10 to 400 ng/mL was developed and validated to support clinical trials using S1P as a biomarker.
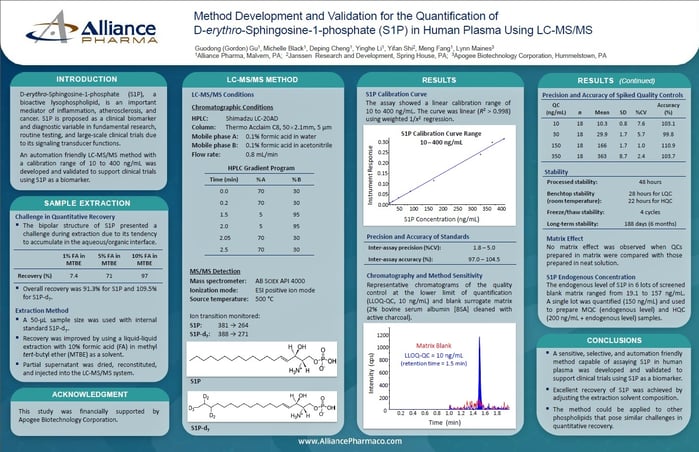
Sample Extraction
Challenge in Quantitative Recovery
The bipolar structure of S1P presented a challenge during extraction due to its tendency to accumulate in the aqueous/organic interface.
Overall recovery was 91.3% for S1P and 109.5% for S1P-d7.
Extraction Method
- A 50-uL sample size was used with internal standard S1P-d7.
- Recovery was improved by using a liquid-liquid extraction with 10% formic acid (FA) in methyl tert-butyl ether (MTBE) as a solvent.
- Partial supernatant was dried, reconstituted, and injected into the LC-MS/MS system.
LC-MS/MS Method
LC-MS/MS Conditions
HPLC: Shimadzu LC-20AD
Column: Thermo Acclaim C8, 50x2.1mm, 5um
Mobile phase A: 0.1% formic acid in water
Mobile phase B: 0.1% formic acid in acetonitrile
Flow rate: 0.8 mL/min
MS/MS Detection
Mass Spectrometer: AB Sciex API 4000
Ionization mode: ESI positive ion mode
Source temperature: 500° C
Ion transition monitered:
S1P: 381 → 264
S1P-d7: 388 → 271
Results
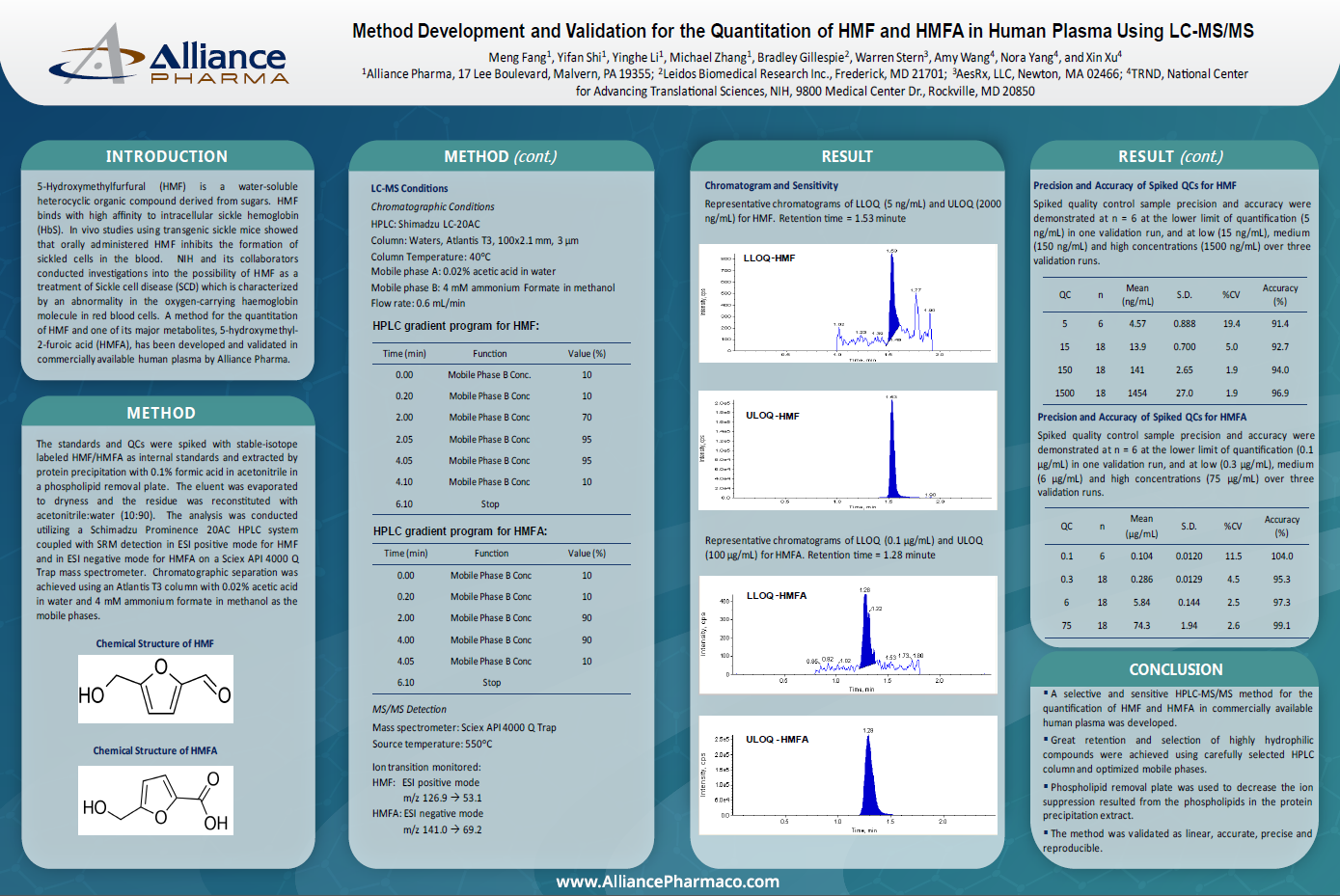
S1P Calibration Curve
The assay showed a linear calibration range of 10 to 400 ng/mL. The curve was linear (R² > 0.998) using weighted 1/x² regression.
Chromatography and Method Sensitivity
Representative chromatograms of the quality control at the lower limit of quantification (LLOQ-QC, 10 ng/mL) and blank surrogate matrix (2% bovine serum albumin [BSA] cleaned with active charcoal).
Matrix Effect
No matrix effect was observed when QCs prepared in matrix were compared with those prepared in neat solution
S1P Endogenous Concentration
The endogenous level of S1P in 6 lots of screened blank matrix ranged from 19.1 to 157 ng/mL. A single lot was quantified (150 ng/mL) and used to prepare MQC (endogenous level) and HQC (200 ng/mL + endogenous level) samples.
Conclusions
- A sensitive, selective, and automation friendly method capable of assaying S1P in human plasma was developed and validated to support clinical trials using S1P as a biomarker.
- Excellent recovery of S1P was achieved by adjusting the extraction solvent composition.
- The method could be applied to other phospholipids that pose similar challenges in quantitative recovery.
Acknowledgement
Guodong (Gordon) Gu, Michelle Black, Deping Cheng, Yinghe Li, Yifan Shi, Meng Fang, Lynn Maines
Alliance Pharma, Malvern, PA; Janssen Research and Development, Spring House, PA; Apogee Biotechnology Corporation, Hummelstown, PA
This study was financially supported by Apogee Biotechnology Corporation.